3 Work Effectively on the UK Biobank Research Analysis Platform (UKB RAP)
3.1 Who This Chapter is For
- You are a bioinformatician who wants to run pipelines or custom software on the UK Biobank data.
- You are a data scientist or epidemiologist and you want to run analyses in RStudio on UKB RAP on either the pheno data or other data fields.
- You are a bioinformatician or data scientist who wants to work in Jupyter notebooks using JupyterLab on UKB RAP.
- You want to explore the UKB Pheno Data using the Cohort Browser platform on UKB RAP.
3.2 Introduction
The UK Biobank Research Analysis Platform (UKB RAP) gives you the ability to work with one of the largest cohorts of people: over 500,000 participants.
If you’re coming from either analyzing data on your own machine or from HPC computing, there are some key differences that you will have to learn.
This chapter attempts to give you the knowledge you need to work successfully on UKB RAP.
3.3 Learning Objectives
- Explain key differences with the UK Biobank Research Analysis Platform (RAP) and the core DNAnexus platform.
- Explain how to process files using built in apps on RAP using Swiss Army Knife.
- Identify two key methods for extracting Pheno Data in a RAP project.
- Explain the two main strategies for listing and accessing files in a RAP project in JupyterLab or RStudio (
dx find dataversus/mnt/project/). - Utilize JupyterLab on RAP reproducibly with Python/R/bash.
- Utilize RStudio on RAP reproducibly.
3.4 What’s Different?
Here are the main differences with UK Biobank and the Core DNAnexus Platform.
- On UKB RAP, you need to apply for access and note what fields are relevant for your work. When the UK Biobank team has reviewed it, you will be assigned an Application ID.
- On UKB RAP, you are limited to sharing projects within your own application ID. All billing information is attached to the application ID, rather than to an organization.
- On UKB RAP, data is dispensed when requested at project creation or refresh. This is both the pheno data (the
dataset) and the Bulk, or file based data. - On UKB RAP, The participant identifiers (
eids) are different across applications because of the psuedonymization process. - On UKB RAP, there is a
Bulk/File Folder that contains many of the files you want to analyze. This includes pVCF files, but also metabolomics data, imaging data, accelerometer data, and proteomics data, as well.
The UK Biobank Community (at https://community.dnanexus.com) is an incredible resource to ask questions, see answers, learn about webinars/tutorials, and understand.
Just know that you’re part of a huge group of researchers who are learning to use RAP and they have probably encountered similar issues to you.
3.5 Application ID and Sharing
When you file an access application with UK Biobank, you need to supply a list of fields that you will use in your research.
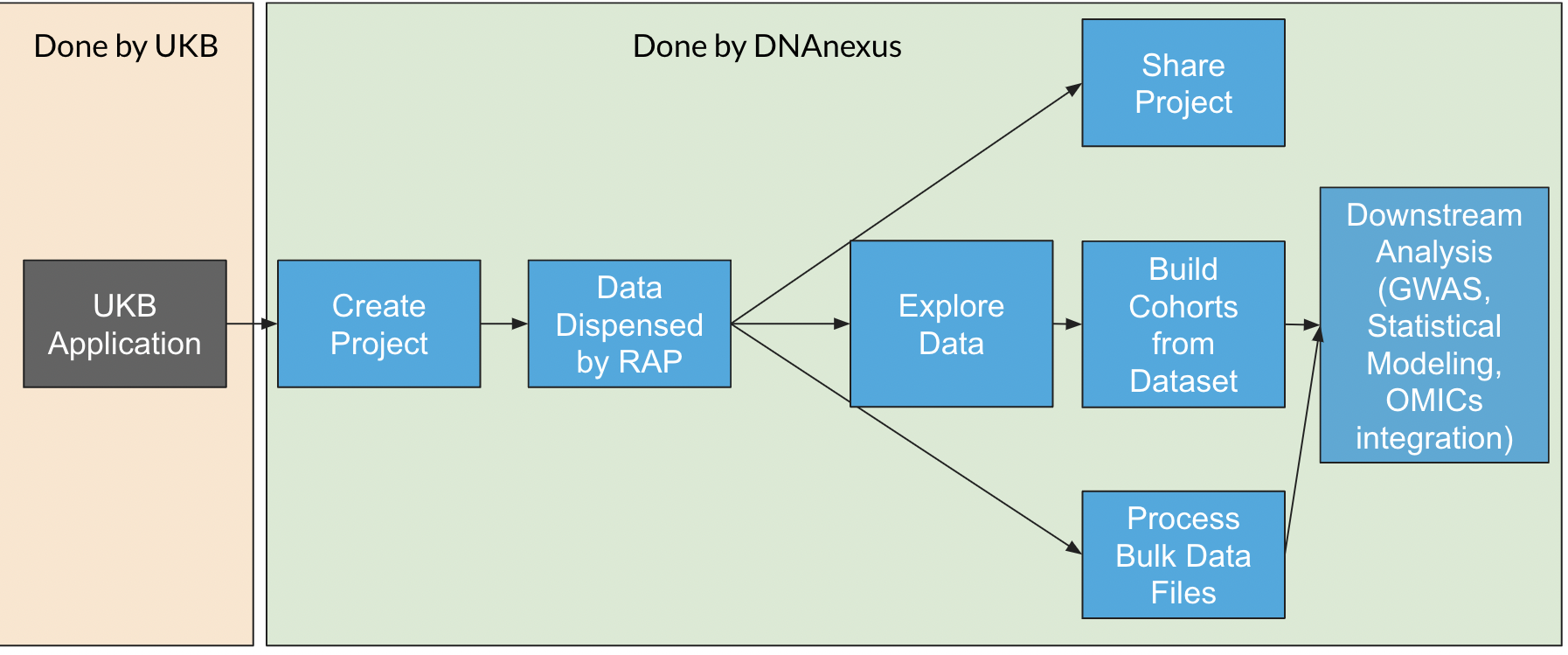
3.6 Data Dispensal
The data dispensal process begins when you create a RAP project within your application ID. When you request data to be dispensed, it may take 20 minutes to several hours for the data to appear in your project, depending on the data fields you have requested in your application.
When data is dispensed, two things happen in your project:
- A
/Bulk/folder is created in your project that contains files associated with data, such as.vcf.gzfiles, or geno data inBGENformat. - A
datasetis created in your project, which contains both the pheno data fields and the processed geno fields. This dataset consists of multiple Spark Databases. This dataset can be browsed using the Cohort Browser if you click on it in your project.
When the data is refreshed on RAP (which may take a few weeks to months following its release on UKB Showcase), you are able to update the dispensed Data.
3.7 Pseudonymization and Participant IDs
Within an application, participant IDs (also known as EIDs) are unique, through the pseudonymization process. This process has been done to ensure security and anonymity of participants.
This means that you cannot join data across applications.
For example, if I’m in application ID 43333 and you’re in application 11111, our EIDs will not correspond because we both have a set of unique EIDs. Our data files are also mapped to these unique IDs.
So if I make a pheno matrix with EIDs and you want to reuse this pheno matrix in your application, it won’t work, because our EIDs don’t match up.
3.8 Pheno/Geno Data
Pheno and Processed Geno Data are stored within multiple Spark databases within your RAP Project. These physical databases are accessed through a single object called the dataset.
There is a built in application called Cohort Browser that will let you explore data fields and derive cohorts by building filters on the data. These cohorts can be shared with others on your application.
Once you have derived cohorts, you can extract the pheno fields by using table exporter or extracting the data.
3.9 Bulk Files
The other difference with UKB RAP is that a lot of the data (image, genomics, etc) are available as files that have been dispensed into your Bulk/ folder.
Here are a few examples of searching for bulk files using dx find data:
3.9.1 Search on Field ID
3.9.2 Search on EID
When you work with the UKB Pheno Data, you will need to find Field IDs for particular fields that you’re interested in. Sometimes there are multiple measurements/instances for Field IDs.
To understand the structure of the data (and what field IDs to grab), I highly recommend using UKB Showcase to browse the fields and search for fields of interest.
3.10 Example Jobs on UKB
For a review of running jobs and useful bash tips, please check out Bash for Bioinformatics, especially the Using
dx runand Batch Processing chapters.
3.11 Extracting Pheno Data from the RAP Dataset
This is a question we get a lot. I’ll explain two ways to extract the pheno data:
- If you are less command-line savvy: Use the Table Exporter App to export the phenotype fields to a CSV file. You have the option to select a decoded file when you run it.
- If you have started to use
dx-toolkit: Use thedx-toolkitcommanddx extract_datasetfrom Python ordx extract_datasetfrom R to extract raw values to a CSV file, and decode the file manually. If your query takes longer than 2 minutes to run, then you will have to start a Spark JupyterLab Cluster and run the SQL query generated bydx extract_datasetwith the--sqloption.
The first thing you need to do is identify the dataset in your project. Make sure that you chose to dispense data in your project (see above), and the dataset should be in the root of your project and begin with dataset-.
You’ll want to grab the record ID for your dataset. You can find it by selecting your dataset in the GUI and clicking the “i” button in the top-left.
3.12 Using JupyterLab on RAP
3.12.1 Using Stata/JupyterLab on RAP
3.13 Using R/RStudio on RAP
One of the nice features of RAP is that you can run R/RStudio on it.
Because there are two kinds of projects, the RAP Project and your RStudio Project, the discussion can be confusing.
I’ll refer to the RAP Project as RAP Storage, and the RStudio Project as a RStudio Project. Hopefully that will make things less ambiguous.
The first thing you’ll notice is that the Files tab in RStudio is isolated from the project storage.
To access data in your RStudio project, you will have to use dx download or dxFUSE to load the data.
The second thing you might notice is that your installed libraries are not installed when you open a session.
These two issues make working with RStudio on RAP different than working with RStudio on your own machine.
Here’s a quick table that talks about the main differences with using RStudio on RAP compared to a local installation of RStudio.
| Feature | Local RStudio | RStudio on RAP |
|---|---|---|
| Accessing Files | Has Access to your local filesystem | Access files in RAP Storage using dxfuse (/mnt/project/myfile.txt/) or using dx download |
| Installing Packages | Use install.packages() |
Init renv in project, then install packages |
| Saving Projects | Save files in project | Use snapshot dx backup-folder capability to save project folder to RAP Storage |
| Opening Projects | Open .rproj file in Project |
Use dx restore-folder to restore project folder, then open .rproj file |
Just because you have closed your RStudio Session does not mean that the job is terminated. Use the red terminate button on the top of the RStudio interface to stop the cloud worker.
3.13.1 Looking at files in your RAP Project
Let’s take a quick look at the files in our /Bulk/ folder. In the RStudio console, we can use list.files() to do this.
Note we put a /mnt/project/ in front of /Bulk/. This is how we access the file system contents using dxFUSE.
list.files("/mnt/project/Bulk/")The response from the console is this:
[1] "Activity" "Brain MRI"
[3] "Carotid Ultrasound" "Electrocardiogram"
[5] "Exome sequences" "Exome sequences_Alternative exome processing"
[7] "Exome sequences_Previous exome releases" "Genotype Results"
[9] "Heart MRI" "Imputation"
[11] "Kidney MRI" "Liver MRI"
[13] "Pancreas MRI" "Protein biomarkers"
[15] "Retinal Optical Coherence Tomography" "Whole Body DXA"
[17] "Whole Body MRI" "Whole genome sequences" Say we want to dive deeper into Exome sequences. We can run:
list.files("/mnt/project/Bulk/Exome sequences")And we’ll see the different formats of the Exome data:
[1] "Exome OQFE CRAM files"
[2] "Exome OQFE variant call files (VCFs)"
[3] "Population level exome OQFE variants, BGEN format - final release"
[4] "Population level exome OQFE variants, BGEN format - interim 450k release"
[5] "Population level exome OQFE variants, PLINK format - final release"
[6] "Population level exome OQFE variants, PLINK format - interim 450k release"
[7] "Population level exome OQFE variants, pVCF format - final release"
[8] "Population level exome OQFE variants, pVCF format - interim 450k release" Within each of these folders is a series of numbered folders that have two digits. The two digits (such as 11) are the first two digits of the EIDs that are in the folder. For example, a file with an EID of 2281131 is going to be in 22/ folder.
There are some hard limits to the number of files that can be in a folder. Thus, each set of 500K files (such as .vcf.gz) is split up into multiple folders with about 10000 files in each.
3.13.2 Reproducible Research using RStudio on RAP
My key piece of advice when running RStudio on RAP is to use the following process:
For starting a project:
- Initialize a new RStudio project.
- Use
renv::init()to initializerenvin your RStudio Project. - Install needed packages to your RStudio Project with
install.packages(). - If you need to work with pheno data, use Table Exporter or
dx extract_datasetto extract it to a CSV file on RAP Storage. - Load data either using
dx download(for smaller batches of files) or dxFUSE (using paths like/mnt/project/myfile.txt).
For working in your project:
- Do your work, saving it as an
Rmarkdownorquartodocument. - Run
renv::snapshot()in your RStudio project. - Use the
dx-toolkitsnapshot functionality indx-toolkitto save (dx backup-folder) your project into RAP Storage. (dx-backup-folder -d /.Backups/my_project.tar.gz)
For example, in an RStudio Project we can run this command in the terminal:
dx-backup-folder -d /.Backups/rstudio_test_project.tar.gzIn this command, we’re backing up the current RStudio Project folder into a file called /.Backups/rstudio_test_project.tar.gz on RAP storage. We’re using the -d (destination) option to do this. This will give us the following response:
Folder . was successfully saved to /.Backups/rstudio_test_project.tar.gz ( file-GBFf5vQJ9Zz93fGk0X8pV2Bf )To resume an RStudio project:
- To resume your work, restore (
dx restore-folder) your RStudio project from RAP Storage. - Open up the
.rprojfile in RStudio to resume your RStudio project. - To install more packages, use
renv::activate(),install.packages(), and thenrenv::snapshot(). - Use
dx backup-folderto save your work.
For example, we can use this command in our terminal to expand our my_project.tar.gz into a project folder called my_project:
dx-restore-folder /.Backups/my_project.tar.gz -d my_projectOnce we click on the .rproj folder in our project, we can resume our work.
If we want to add new packages, we can run the following in the RStudio console:
renv::activate()Then we can install.packages() as usual.
Then we do our work, and then when we’re done, we’ll use:
`renv::snapshot()
to save our packages into our Project. Then we can use dx-backup-folder as usual.
renv?
renv is a R Package that implements virtual environments for your RStudio project.
In short, a virtual environment allows you to completely reproduce the software environment (all the packages with their version numbers) in your project.
How does renv work? Each RStudio Project has its own library of packages, which is modified as you install packages in your project. This is also handy in that you can work with multiple projects that each have their own version of a package.
This seems to be a lot of extra work. Why do it? Well, this process alleviates a lot of complaints about working with a cloud instance of RStudio:
- It allows you to resume work on a RStudio project without having to reinstall packages or redownload data. Everything is saved in the project folder, which is then backed up to RAP Storage.
- It ties your packages to version numbers. Sharing an RStudio Project with
renvinitialized means that your collaborators can install an identical software environment and reproduce your work.