----------------------------------------
Global fields:
None
----------------------------------------
Column fields:
's': str
'sample_qc': struct {
dp_stats: struct {
mean: float64,
stdev: float64,
min: float64,
max: float64
},
gq_stats: struct {
mean: float64,
stdev: float64,
min: float64,
max: float64
},
call_rate: float64,
n_called: int64,
n_not_called: int64,
n_filtered: int64,
n_hom_ref: int64,
n_het: int64,
n_hom_var: int64,
n_non_ref: int64,
n_singleton: int64,
n_snp: int64,
n_insertion: int64,
n_deletion: int64,
n_transition: int64,
n_transversion: int64,
n_star: int64,
r_ti_tv: float64,
r_het_hom_var: float64,
r_insertion_deletion: float64
}
----------------------------------------
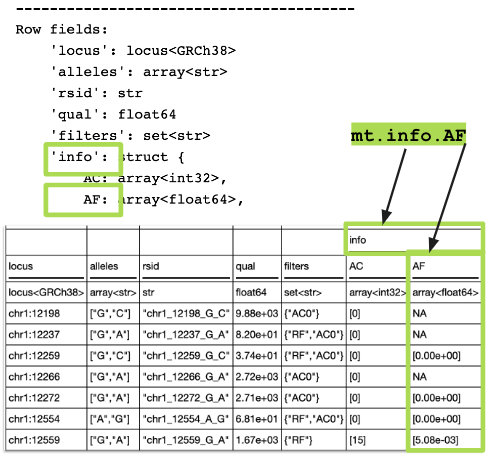
Row fields:
'locus': locus<GRCh38>
'alleles': array<str>
'rsid': str
'qual': float64
'filters': set<str>
'info': struct {
AC: array<int32>,
AF: array<float64>,
AN: int32,
BaseQRankSum: float64,
ClippingRankSum: float64,
DB: bool,
DP: int32,
FS: float64,
InbreedingCoeff: float64,
MQ: float64,
MQRankSum: float64,
QD: float64,
ReadPosRankSum: float64,
SOR: float64,
VQSLOD: float64,
VQSR_culprit: str,
VQSR_NEGATIVE_TRAIN_SITE: bool,
VQSR_POSITIVE_TRAIN_SITE: bool,
GQ_HIST_ALT: array<str>,
DP_HIST_ALT: array<str>,
AB_HIST_ALT: array<str>,
GQ_HIST_ALL: str,
DP_HIST_ALL: str,
AB_HIST_ALL: str,
AC_AFR: array<int32>,
AC_AMR: array<int32>,
AC_ASJ: array<int32>,
AC_EAS: array<int32>,
AC_FIN: array<int32>,
AC_NFE: array<int32>,
AC_OTH: array<int32>,
AC_SAS: array<int32>,
AC_Male: array<int32>,
AC_Female: array<int32>,
AN_AFR: int32,
AN_AMR: int32,
AN_ASJ: int32,
AN_EAS: int32,
AN_FIN: int32,
AN_NFE: int32,
AN_OTH: int32,
AN_SAS: int32,
AN_Male: int32,
AN_Female: int32,
AF_AFR: array<float64>,
AF_AMR: array<float64>,
AF_ASJ: array<float64>,
AF_EAS: array<float64>,
AF_FIN: array<float64>,
AF_NFE: array<float64>,
AF_OTH: array<float64>,
AF_SAS: array<float64>,
AF_Male: array<float64>,
AF_Female: array<float64>,
GC_AFR: array<int32>,
GC_AMR: array<int32>,
GC_ASJ: array<int32>,
GC_EAS: array<int32>,
GC_FIN: array<int32>,
GC_NFE: array<int32>,
GC_OTH: array<int32>,
GC_SAS: array<int32>,
GC_Male: array<int32>,
GC_Female: array<int32>,
AC_raw: array<int32>,
AN_raw: int32,
AF_raw: array<float64>,
GC_raw: array<int32>,
GC: array<int32>,
Hom_AFR: array<int32>,
Hom_AMR: array<int32>,
Hom_ASJ: array<int32>,
Hom_EAS: array<int32>,
Hom_FIN: array<int32>,
Hom_NFE: array<int32>,
Hom_OTH: array<int32>,
Hom_SAS: array<int32>,
Hom_Male: array<int32>,
Hom_Female: array<int32>,
Hom_raw: array<int32>,
Hom: array<int32>,
STAR_AC: int32,
STAR_AC_raw: int32,
STAR_Hom: int32,
POPMAX: array<str>,
AC_POPMAX: array<int32>,
AN_POPMAX: array<int32>,
AF_POPMAX: array<float64>,
DP_MEDIAN: array<int32>,
DREF_MEDIAN: array<float64>,
GQ_MEDIAN: array<int32>,
AB_MEDIAN: array<float64>,
AS_RF: array<float64>,
AS_FilterStatus: array<str>,
AS_RF_POSITIVE_TRAIN: array<int32>,
AS_RF_NEGATIVE_TRAIN: array<int32>,
AC_AFR_Male: array<int32>,
AC_AMR_Male: array<int32>,
AC_ASJ_Male: array<int32>,
AC_EAS_Male: array<int32>,
AC_FIN_Male: array<int32>,
AC_NFE_Male: array<int32>,
AC_OTH_Male: array<int32>,
AC_SAS_Male: array<int32>,
AC_AFR_Female: array<int32>,
AC_AMR_Female: array<int32>,
AC_ASJ_Female: array<int32>,
AC_EAS_Female: array<int32>,
AC_FIN_Female: array<int32>,
AC_NFE_Female: array<int32>,
AC_OTH_Female: array<int32>,
AC_SAS_Female: array<int32>,
AN_AFR_Male: int32,
AN_AMR_Male: int32,
AN_ASJ_Male: int32,
AN_EAS_Male: int32,
AN_FIN_Male: int32,
AN_NFE_Male: int32,
AN_OTH_Male: int32,
AN_SAS_Male: int32,
AN_AFR_Female: int32,
AN_AMR_Female: int32,
AN_ASJ_Female: int32,
AN_EAS_Female: int32,
AN_FIN_Female: int32,
AN_NFE_Female: int32,
AN_OTH_Female: int32,
AN_SAS_Female: int32,
AF_AFR_Male: array<float64>,
AF_AMR_Male: array<float64>,
AF_ASJ_Male: array<float64>,
AF_EAS_Male: array<float64>,
AF_FIN_Male: array<float64>,
AF_NFE_Male: array<float64>,
AF_OTH_Male: array<float64>,
AF_SAS_Male: array<float64>,
AF_AFR_Female: array<float64>,
AF_AMR_Female: array<float64>,
AF_ASJ_Female: array<float64>,
AF_EAS_Female: array<float64>,
AF_FIN_Female: array<float64>,
AF_NFE_Female: array<float64>,
AF_OTH_Female: array<float64>,
AF_SAS_Female: array<float64>,
GC_AFR_Male: array<int32>,
GC_AMR_Male: array<int32>,
GC_ASJ_Male: array<int32>,
GC_EAS_Male: array<int32>,
GC_FIN_Male: array<int32>,
GC_NFE_Male: array<int32>,
GC_OTH_Male: array<int32>,
GC_SAS_Male: array<int32>,
GC_AFR_Female: array<int32>,
GC_AMR_Female: array<int32>,
GC_ASJ_Female: array<int32>,
GC_EAS_Female: array<int32>,
GC_FIN_Female: array<int32>,
GC_NFE_Female: array<int32>,
GC_OTH_Female: array<int32>,
GC_SAS_Female: array<int32>,
Hemi_AFR: array<int32>,
Hemi_AMR: array<int32>,
Hemi_ASJ: array<int32>,
Hemi_EAS: array<int32>,
Hemi_FIN: array<int32>,
Hemi_NFE: array<int32>,
Hemi_OTH: array<int32>,
Hemi_SAS: array<int32>,
Hemi: array<int32>,
Hemi_raw: array<int32>,
STAR_Hemi: int32
}
----------------------------------------
Entry fields:
'GT': call
'AD': array<int32>
'DP': int32
'GQ': int32
'PL': array<int32>
----------------------------------------
Column key: ['s']
Row key: ['locus', 'alleles']
----------------------------------------